Retinoblastoma
Retinoblastoma is the most common malignant tumor in the eye(s) of children. It is caused by genetic changes known as mutations. Such mutations are nothing unusual – every child has around three new mutations in its genetic material.
However, retinoblastoma only develops when a mutation occurs in one specific gene of the approximately 30,000 genes in our cells and this gene loses its function as a result. This special gene is called the retinoblastoma gene, or RB1 gene for short, and is located on chromosome 13. Every person has two copies of the RB1 gene, one from the mother and one from the father. Retinoblastoma only develops when both copies of the RB1 gene are mutated.
This tumor can only develop from certain cells in a child’s eye. This means that mutations of the RB1 gene in other cells of the body do not result in retinoblastoma. Also for this reason this tumor is rare: Only around one in 15,000 children develop the disease.
Some patients with retinoblastoma have a hereditary increased risk of this tumor because one of the two copies of the RB1 gene is also mutated in all cells outside the tumor. Children of a patient with hereditary retinoblastoma are equally likely to either inherit the normal copy and then have no increased risk of retinoblastoma or they inherit the mutated copy and then usually also develop retinoblastoma. In families, relatives with hereditary retinoblastoma can be identified by predictive genetic testing before the tumor occurs. In these cases the course of the disease can be improved by early tumor detection examinations.
Stem cell model for retinoblastoma
Biallelic inactivation of the tumor suppressor gene RB1 results in the formation of retinoblastoma, a childhood eye cancer. Mouse models or established retinoblastoma cell lines are inadequate for studying early stages of tumorigenesis, either because they are not applicable to humans or represent later stages of retinoblastoma. To address this gap, our research focuses on establishing a human stem cell-based model for retinoblastoma.
We have established a protocol for the differentiation of human embryonic stem cells into 3D retinal organoids. Using this protocol, all seven retinal cell types develop in the same layering and order observed during human eye development. One of these cell types are cone photoreceptors, the presumed cells-of-origin of retinoblastoma.
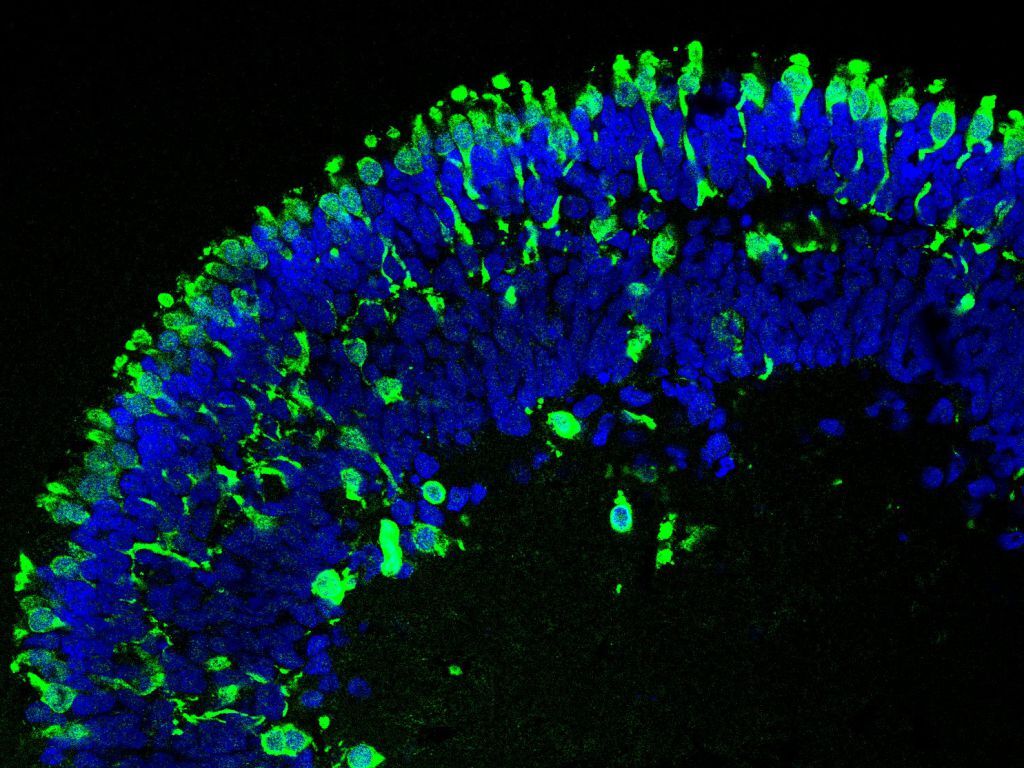
By using the CRISPR/Cas9 system, we introduced mutations into the RB1 gene in an embryonic stem cell line and differentiated the generated cells into retinal organoids. When comparing wildtype (no RB1 mutation), heterozygous (one RB1 allele mutated) and knockout (both RB1 allele mutated) retinal organoids, we found that wildtype and heterozygous organoids were similar in their structure and cellular composition. However, retinal organoids with biallelic RB1 inactivation showed a lower abundance of certain retinal cell types and a structural impairment, as they began to dissociate at later stages of differentiation. Additionally, we observed persistent proliferation in cone photoreceptors, consistent with their proposed role as the cells-of-origin in retinoblastoma (Figure 1).

At day 152 of differentiation the structure of RB1 wildtype (RB1wt) and heterozygous (RB1het) organoids is similar. Immature (RXRγ) and mature cone photoreceptors (ARR3), both shown in green, are located in the outer layer of the organoids. In these organoids proliferating cells (Ki67), stained in red, were rarely found. Only in organoids with biallelic inactivation of RB1 (RB1ko) proliferating cone photoreceptors (positive for RXRγ/ARR3 (green) and Ki67 (red)), indicated by the arrows, can be found. Nuclei are stained in blue. Scale bar: 50 µm.
Figure was adapted and modified from Kanber et al, 2022.
The overall gene expression profile of retinal organoids with biallelic RB1 inactivation closely resembles that of retinoblastoma tumors (Figure 2A), whereas wildtype and heterozygous organoids more closely match fetal retina samples (Figure 2B). Taken together, the organoids in our model exhibit certain characteristics of retinoblastoma, providing a valid human model which enables future research on this disease.

(A) 510 retinoblastoma signature genes were determined from publically available data (RB signature). Retinal organoids with biallelic inactivation of RB1 develop an RB signature from d35 to d152.
(B) The gene expression between fetal retina samples and retinoblastoma tumors was compared. Wildtype(RB1wt) and heterozygous (RB1het) retinal organoids were more similar to fetal retina samples whereas organoids with biallelic inactivation of RB1 (RB1ko) were more comparable to retinoblastoma tumors.
Figure was adapted and modified from Kanber et al, 2022.
Contact person/contact details
Dr. rer. nat.
Deniz Kanber
Dr. rer. nat.
Julia Wöstefeld
Selected publications from the last years
Kanber, D., Woestefeld, J., Döpper, H., Bozet, M., Brenzel, A., Altmüller, J., Kilpert, F., Lohmann, D., Pommerenke, C., & Steenpass, L. (2022).
RB1-Negative Retinal Organoids Display Proliferation of Cone Photoreceptors and Loss of Retinal Differentiation
Cancers, 14(9), 2166. https://doi.org/10.3390/cancers14092166
Döpper, H., Menges, J., Bozet, M., Brenzel, A., Lohmann, D., Steenpass, L., & Kanber, D. (2020).
Differentiation Protocol for 3D Retinal Organoids, Immunostaining and Signal Quantitation
Current protocols in stem cell biology, 55(1), e120. https://doi.org/10.1002/cpsc.120
Döpper, H., Horstmann, M., Menges, J., Bozet, M., Kanber, D., & Steenpass, L. (2020).
Biallelic and monoallelic deletion of the RB1 promoter in six isogenic clonal H9 hESC lines
Stem cell research, 45, 101779. https://doi.org/10.1016/j.scr.2020.101779
Menges, J., Cremanns, M., & Steenpass, L. (2019).
Generation of two H1 hESC sublines carrying deletions of RB1 exon 1/promoter in heterozygous or compound heterozygous state
Stem cell research, 39, 101517. https://doi.org/10.1016/j.scr.2019.101517
Schipper, L., Kanber, D., & Steenpass, L. (2018).
Generation of heterozygous and homozygous hESC H9 sublines carrying inactivating mutations in RB1
Stem cell research, 33, 41–45. https://doi.org/10.1016/j.scr.2018.09.016
Former group members
- Prof. Dr. rer. nat. Laura Steenpaß
- Dr. rer. nat. Hannah Döpper
- Morgane Bozet, M.Sc.
- Leonie Brochhagen, née Schipper, B.Sc.