Retinoblastom
Das Retinoblastom ist der häufigste bösartige Tumor im Auge von Kindern. Es entsteht durch genetische Veränderungen, die als Mutationen bezeichnet werden. Solche Mutationen sind nichts Ungewöhnliches – jedes Kind hat etwa drei neue Mutationen in seiner Erbsubstanz.
Ein Retinoblastom entwickelt sich aber nur dann, wenn eine Mutation in einem bestimmten der etwa 30.000 Genen in unseren Zellen auftritt und dieses Gen dadurch seine Funktion verliert. Dieses spezielle Gen heißt Retinoblastom-Gen, kurz RB1-Gen, und befindet sich auf Chromosom 13. Jeder Mensch hat zwei Kopien des RB1-Gens, eine von der Mutter und eine vom Vater. Ein Retinoblastom entsteht erst, wenn beide Kopien des RB1-Gens mutiert sind.
Dieser Tumor kann sich nur aus bestimmten Zellen im Auge eines Kindes entwickeln. Das bedeutet, dass Mutationen des RB1-Gens in anderen Körperzellen nicht zu einem Retinoblastom führen. Auch deshalb ist dieser Tumor selten: Nur etwa eines von 15.000 Kindern erkrankt daran.
Einige Patienten mit Retinoblastom haben ein erblich erhöhtes Risiko für diesen Tumor, weil eine der beiden Kopien des RB1-Gens auch in allen Zellen außerhalb des Tumors mutiert ist. Kinder eines Patienten mit erblichem Retinoblastom erben mit gleicher Wahrscheinlichkeit entweder die normale Kopie und haben dann kein erhöhtes Retinoblastom-Risiko oder sie erben die mutierte Kopie und erkranken dann meist auch an einem Retinoblastom. In Familien können Angehörige mit erblichem Retinoblastom schon vor dem Auftreten des Tumors durch genetische Untersuchungen erkannt werden. Bei diesen kann der Krankheitsverlauf durch Tumor-Früherkennungsuntersuchungen verbessert werden.
Stammzellmodell für das Retinoblastom
Die biallelische Inaktivierung des Tumorsuppressorgens RB1 führt zur Entstehung eines Retinoblastoms, ein Augentumor im Kindesalter. Mausmodelle oder Retinoblastom-Zelllinien sind für die Untersuchung der frühen Stadien der Tumorentstehung nicht geeignet, da sie entweder nicht auf den Menschen übertragbar sind oder spätere Stadien der Retinoblastomentwicklung repräsentieren. Um diese Lücke zu schließen, konzentriert sich unsere Forschung auf die Etablierung eines humanen, stammzellbasierten Modells für das Retinoblastom.
Wir haben ein Protokoll zur Differenzierung humaner embryonaler Stammzellen in 3D-retinale Organoide entwickelt. Mithilfe dieses Protokolls entwickeln sich alle sieben retinale Zelltypen in derselben Anordnung und Reihenfolge, wie sie während der Entwicklung des menschlichen Auges beobachtet werden. Einer dieser Zelltypen sind die Zapfen-Photorezeptoren, die als Ursprungszellen des Retinoblastoms gelten.
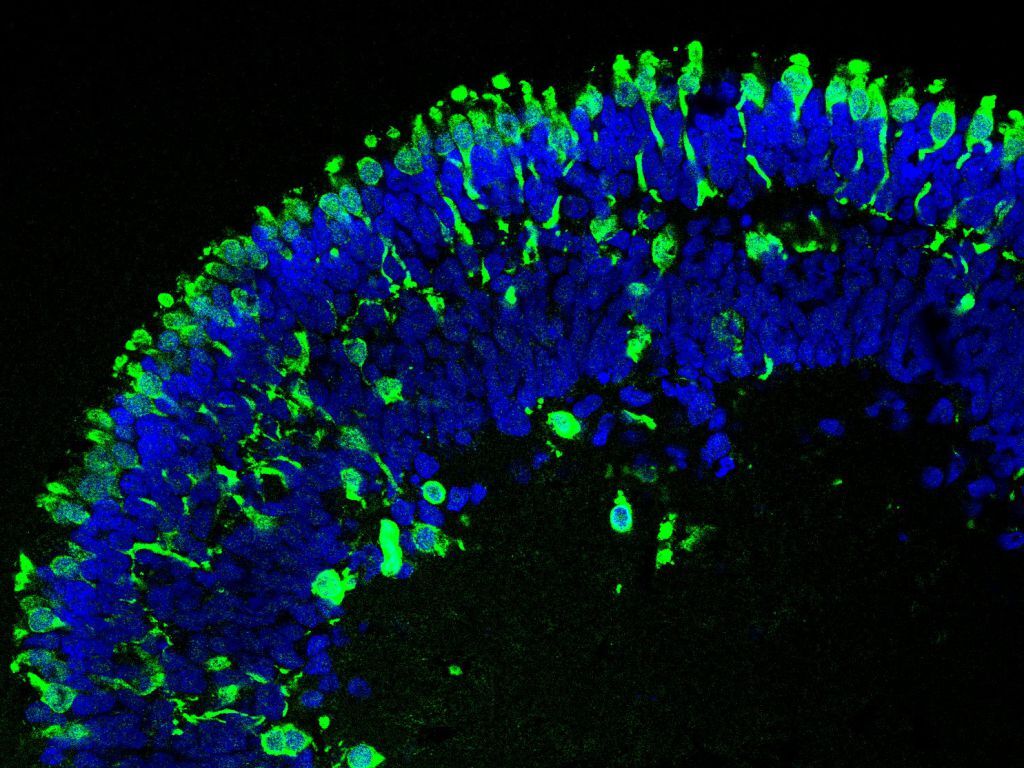
Durch den Einsatz des CRISPR/Cas9-Systems haben wir Mutationen im RB1-Gen in einer embryonalen Stammzelllinie eingebaut und die daraus entstandenen Zellen in retinale Organoide differenziert. Beim Vergleich von Wildtyp-Organoiden (ohne RB1 Mutation), heterozygoten Organoiden (ein RB1 Allel mutiert) und knockout Organoiden (beide RB1 Allele mutiert) stellte sich heraus, dass sich Wildtyp- und heterozygote Organoide in ihrer Struktur und Zellzusammen-setzung ähneln. Retinale Organoide mit biallelischer RB1 Inaktivierung zeigten jedoch eine geringere Anzahl bestimmter retinaler Zelltypen und strukturelle Veränderungen, da die Organoide in späteren Stadien der Differenzierung begannen zu zerfallen. Zudem konnte eine anhaltende Proliferation der Zapfen-Photorezeptoren festgestellt werden, was ihre vermutete Rolle als Ursprungszellen des Retinoblastoms unterstützt (Abbildung 1).

An Tag 152 der Differenzierung ist die Struktur von RB1-Wildtyp (RB1wt) und heterozygoten (RB1het) Organoiden vergleichbar. Unreife (RXRγ) und reife Zapfen-Photorezeptoren (ARR3), beide in grün dargestellt, befinden sich in der äußeren Schicht der Organoide. Proliferierende Zellen (Ki67), rot gefärbt, waren in diesen Organoiden nur vereinzelt zu finden. Nur in Organoiden mit biallelischer Inaktivierung von RB1 (RB1ko) finden sich proliferierende Zapfen-Photorezeptoren (positiv für RXRγ/ARR3 (grün) und Ki67 (rot)), gekennzeichnet durch die Pfeile. Die Zellkerne sind blau gefärbt. Maßstab: 50 µm.
Die Abbildung wurde adaptiert und modifiziert von Kanber et al, 2022.
Das Gesamtprofil der Genexpression von retinalen Organoiden mit biallelischer RB1 Inaktivierung ähnelt stark dem von Retinoblastom-Tumoren (Abbildung 2A), während Wildtyp- und heterozygote Organoide eher den fetalen Retinaproben entsprechen (Abbildung 2B). Insgesamt zeigen die Organoide in unserem Modell bestimmte Merkmale des Retinoblastoms und bieten ein validiertes menschliches Modell, das zukünftige Forschungen zu dieser Krankheit ermöglicht.

(A) Aus öffentlich zugänglichen Daten wurden 510 Retinoblastom-Signaturgene ermittelt (RB-Signatur). Retinale Organoide mit biallelischer Inaktivierung von RB1 entwickeln eine RB-Signatur von d35 zu d152.
(B) Die Genexpression zwischen fetalen Retinaproben und Retinoblastom-Tumoren wurde verglichen. Wildtyp (RB1wt) und heterozygote (RB1het) retinale Organoide ähnelten eher fetalen Retinaproben, während Organoide mit biallelischer Inaktivierung von RB1 (RB1ko) eher mit Retinoblastom-Tumoren vergleichbar waren.
Die Abbildung wurde adaptiert und modifiziert von Kanber et al, 2022.
Ansprechpartner/Kontakt
Dr. rer. nat.
Deniz Kanber
Dr. rer. nat.
Julia Wöstefeld
Ausgewählte Publikationen der letzten Jahre
Kanber, D., Woestefeld, J., Döpper, H., Bozet, M., Brenzel, A., Altmüller, J., Kilpert, F., Lohmann, D., Pommerenke, C., & Steenpass, L. (2022)
RB1-Negative Retinal Organoids Display Proliferation of Cone Photoreceptors and Loss of Retinal Differentiation
Cancers, 14(9), 2166. https://doi.org/10.3390/cancers14092166
Döpper, H., Menges, J., Bozet, M., Brenzel, A., Lohmann, D., Steenpass, L., & Kanber, D. (2020)
Differentiation Protocol for 3D Retinal Organoids, Immunostaining and Signal Quantitation
Current protocols in stem cell biology, 55(1), e120. https://doi.org/10.1002/cpsc.120
Döpper, H., Horstmann, M., Menges, J., Bozet, M., Kanber, D., & Steenpass, L. (2020)
Biallelic and monoallelic deletion of the RB1 promoter in six isogenic clonal H9 hESC lines
Stem cell research, 45, 101779. https://doi.org/10.1016/j.scr.2020.101779
Menges, J., Cremanns, M., & Steenpass, L. (2019)
Generation of two H1 hESC sublines carrying deletions of RB1 exon 1/promoter in heterozygous or compound heterozygous state.
Stem cell research, 39, 101517. https://doi.org/10.1016/j.scr.2019.101517
Schipper, L., Kanber, D., & Steenpass, L. (2018)
Generation of heterozygous and homozygous hESC H9 sublines carrying inactivating mutations in RB1
Stem cell research, 33, 41–45. https://doi.org/10.1016/j.scr.2018.09.016
Ehemalige Mitarbeiter
- Prof. Dr. rer. nat. Laura Steenpaß
- Dr. rer. nat. Hannah Döpper
- Morgane Bozet, M.Sc.
- Leonie Brochhagen, geb. Schipper, B.Sc.