
Neurogenetics
Molecular genetics of neurogenetic
and neurodevelopmental disorders
Main research areas: Neurogenetics
Neurogenetic disorders are a clinically heterogeneous group of conditions characterized by an alteration or dysfunction of the central nervous system, resulting from a change encoded in the genome of affected individuals. Most of these conditions are individually rare but collectively frequent (>1-5% of the population), manifesting during development (neurodevelopmental disorders) or during later life (neurological disorders). Currently, genetic alterations in approximately 2,500 genes have been linked to human disorders affecting the brain and collectively explain approximately 50% of patients with neurological disorders with a supposed genetic etiology.
My group has worked on the identification of genetic factors involved in neurodevelopmental (epilepsy, intellectual disability, autism spectrum disorders, brain malformations) and neurodegenerative disorders (hereditary spastic paraplegia, cerebellar ataxia…) since 2002. We have identified or contributed to the identification of approximately 50 new genetic conditions. Examples of disorders we identified include neurodevelopmental disorders caused by pathogenic variants in HCN1, HNRNPU and KCNN2, mirror movements (RAD51), leukoencephalopathy with ataxia (CLCN2) and agenesis of the corpus callosum (DCC). More recently, our group significantly contributed to the identification of noncoding repeat expansions in familial adult myoclonic epilepsy (FAME) and spinocerebellar ataxias (SCAs). We also have a strong interest in establishing genotype-phenotype correlations and understanding the mechanisms underlying incomplete penetrance and variable expressivity. We are member or partner of several international research consortia: IRC5 (https://www.irc5.org/), the SPATAX network, FAME consortium, EuroEPINOMICS RES /EpiCare and SOLVE-RD.
Ongoing projects
We aim to identify new genes and uncover novel mechanisms underlying neurological and neurodevelopmental disorders, with a current focus on noncoding regions and complex (repeated) regions. We use a range of complementary approaches that make use of advanced technologies and bioinformatics tools. We combine short-read genome sequencing and long-read technologies, including nanopore sequencing and optical genome mapping, with multi-omics approaches (RNA sequencing, proteomics…) to detect pathogenic variants missed by routine diagnostics and to discover new genetic diseases. We also apply machine learning to predict gene-disease associations (Leitão et al. Nat Commun, 2024). For selected genetic disorders, we also develop cellular models to explore the underlying pathophysiological mechanisms. Specific projects are listed below.
Tandem repeats are unstable, polymorphic variations abundantly present in human genomes. Their expansion is a well-known pathological process involved in ~60 neurological disorders. The contribution of repeat expansions to the missing heritability of neurological disorders remains largely unknown due to methodological difficulties in mapping reads filled with repeats and estimating repeat lengths on both allele using current next generation sequencing technologies. The goal of this project is to systematically assess the contribution of repeat expansions in patients with unsolved neurological disorders. Our strategy consists in sequentially combining short-read sequencing and long-read Oxford nanopore technologies. We use state-of-the-art bioinformatic pipelines to perform case-control comparisons and outlier analyses and detect repeat expansions.

Post-doctoral researchers: Elsa Leitão, Christopher Schröder
Familial adult myoclonic epilepsy (FAME) is a rare autosomal dominant disorder characterized by myoclonic tremor and seizures. Recently, the same intronic expansion composed of TTTTA/TTTCA repeats in seven different genes have been identified as the cause of FAME. We have gathered a unique collection of families and have identified expansions at two new loci on chromosomes 2 (STARD7) and 5 (MARCHF6). The goal of this project is to dissect the mechanisms by which TTTCA repeat expansions occur and lead to this enigmatic disorder. In particular, we are developing cellular and animal models harboring FAME expansions by generating IPSC that will be differentiated into cortical neurons. Our aim is to clarify the impact of expansions on gene expression and RNA foci formation.

Post-doctoral researcher: Theresa Kühnel
RBMX is an X-linked gene (in mammals) that encodes the RNA-binding hnRNP-G protein, a protein regulating splicing as part of the spliceosome machinery. Pathogenic variants in RBMX are associated with a neurodevelopmental disorder affecting hemizygous males. RBMX has several intronless retrocopies located on autosomes in mammalian genomes. The aims of this project are to investigate the mechanisms by which novel human mutations identified in unrelated families lead to brain disorders and dissect the molecular mechanisms by which RBMX and its functional retrocopies (RBMXL1) contribute to brain development in both human and mouse using complementary cellular human and in vivo mouse models multiomics (RNAseq and proteomics) analyses.
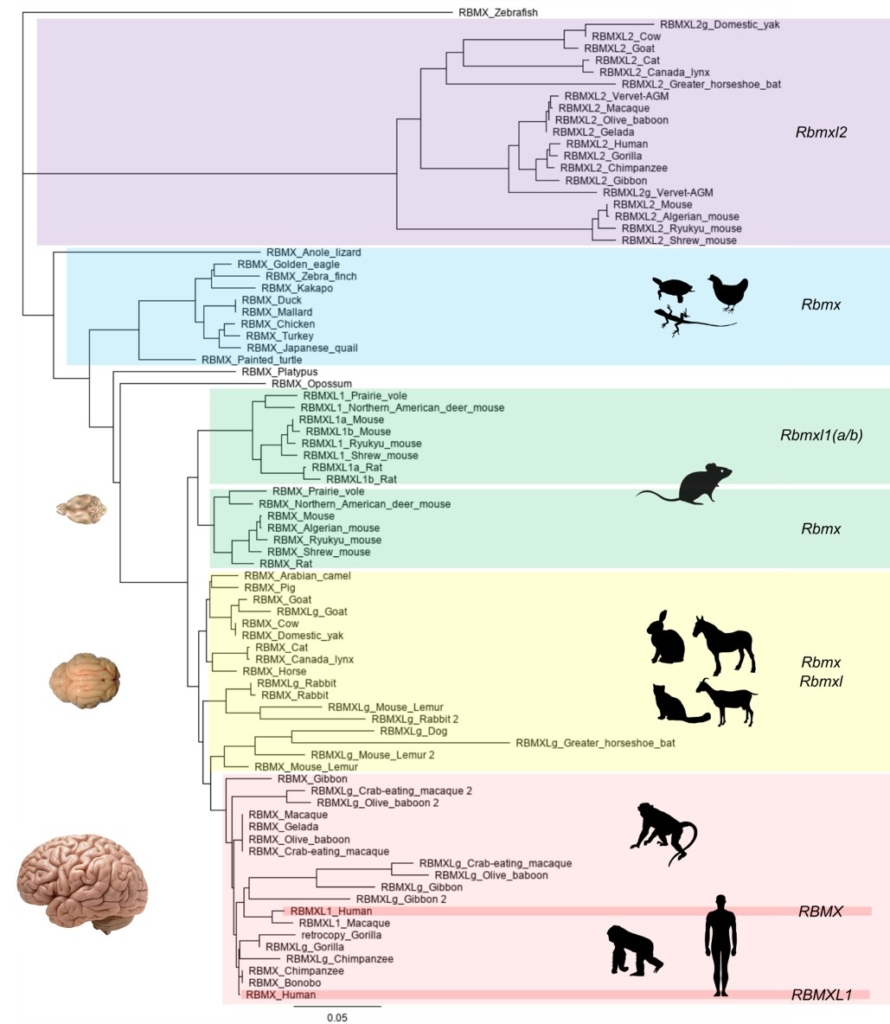
PhD Student: Carolin Mattausch
Project in collaboration with Dr. Juliette Godin (IGBMC, Strasbourg)
Variants in the RNU4-2 gene, encoding the small nuclear RNA (snRNA) U4 involved in the spliceosome, have emerged as a major cause of neurodevelopmental disorders (Chen at al. Nature, 2024). The aims of this project are to 1) collect genetic and clinical data of families with pathogenic variants in RNU4-2 and other snRNA genes; 2) to confirm pathogenicity of variants by studying their segregation within families and perform functional experiments; 3) to dissect the molecular mechanisms by which variants in RNU4-2 and other snRNA genes lead to neurodevelopmental disorders by performing RNA sequencing experiments; and 4) to study ratios of mutant versus normal snRNA U4 but also U1, U2, U5 and U6, that have complementary functions within the spliceosome.

Post-doctoral researcher: Elsa Leitão
Project in collaboration with Dr. Caroline Nava (ICM, Paris) and Prof. Julien Thevenon (Grenoble)
Group members
Prof. Dr. rer. nat.
Christel Depienne
Dr. rer. nat.
Elsa Leitão
M. Sc.
Carolin Mattausch
Sabine Kaya
Dr. med.
Friedrich Erdlenbruch
Adrian Knezovic
B. Sc.
Robin Paluch
Contact person/contact details
Prof. Dr. rer. nat.
Christel Depienne
Neurogenetik
Selected publications
Mohren L*, Erdlenbruch F*, Leitão E*, Kilpert*, G Hönes GS, Kaya S, Schröder C, Thieme A, Sturm M, Park J, Schlüter A, Ruiz M, Morales de la Prida M, Casasnovas C, Becker K, Roggenbuck U, Pechlivanis S, Kaiser FJ, Synofzik M, Wirth T, Anheim M, Haack TB, Lockhart PJ, Jöckel KH, Pujol A, Klebe S, Timmann D, Depienne C. Identification and characterisation of pathogenic and non-pathogenic FGF14 repeat expansions. Nat Commun. 2024 In press.
Chen Y, Dawes R, Kim HC, Ljungdahl A, Stenton SL, Walker S, Lord J, Lemire G, Martin-Geary AC, Ganesh VS, Ma J, Ellingford JM, Delage E, D’Souza EN, Dong S, Adams DR, […] Zocche D, Rubenstein JL, Markenscoff-Papadimitriou E, Fica SM, Baralle D, Depienne C, MacArthur DG, Howson JMM, Sanders SJ, O’Donnell-Luria A, Whiffin N. De novo variants in the RNU4-2 snRNA cause a frequent neurodevelopmental syndrome. Nature. 2024 Jul 11. PMID: 38991538
Schuermans N*, El Chehadeh S*, Hemelsoet D*, Gautheron J*, Vantyghem MC, Nouioua S, Tazir M, Vigouroux C, Auclair M, Bogaert E, Dufour S, Okawa F, Hilbert P, Van Doninck N, Taquet MC, Rosseel T, De Clercq G, Debackere E, Van Haverbeke C, Cherif FR, Urtizberea JA, Chanson JB, Funalot B, Authier FJ, Kaya S, Terryn W, Callens S, Depypere B, Van Dorpe J; Program for Undiagnosed Diseases (UD-PrOZA); Poppe B, Impens F, Mizushima N, Depienne C, Jéru I*, Dermaut B*. Loss of phospholipase PLAAT3 causes a mixed lipodystrophic and neurological syndrome due to impaired PPARγ signaling. Nat Genet. 2023 Nov;55(11):1929-1940. PMID: 37919452
Roos A, van der Ven PFM, Alrohaif H, Kölbel H, Heil L, Della Marina A, Weis J, Aßent M, Beck-Wödl S, Barresi R, Töpf A, O’Connor K, Sickmann A, Kohlschmidt N, El Gizouli M, Meyer N, Daya N, Grande V, Bois K, Kaiser FJ, Vorgerd M, Schröder C, Schara-Schmidt U, Gangfuss A, Evangelista T, Röbisch L, Hentschel A, Grüneboom A, Fuerst DO, Kuechler A, Tzschach A*, Depienne C*, Lochmüller H*. Bi-allelic variants of FILIP1 cause congenital myopathy, dysmorphism and neurological defects. Brain. 2023 Oct 3;146(10):4200-4216. PMID: 37163662
Leitão E, Schröder C, Parenti I, Dalle C, Rastetter A, Kühnel T, Kuechler A, Kaya S, Gérard B, Schaefer E, Nava C, Drouot N, Engel C, Piard J, Duban-Bedu B, Villard L, Stegmann APA, Vanhoutte EK, Verdonschot JAJ, Kaiser FJ, Tran Mau-Them F, Scala M, Striano P, Frints SGM, Argilli E, Sherr EH, Elder F, Buratti J, Keren B, Mignot C, Héron D, Mandel JL, Gecz J, Kalscheuer VM, Horsthemke B, Piton A, Depienne C. Systematic analysis and prediction of genes associated with monogenic disorders on human chromosome X. Nat Commun. 2022 Nov 2;13(1):6570. PMID: 36323681
Parenti I*, Lehalle D*, Nava C, Torti E, Leitão E, Person R, Mizuguchi T, Matsumoto N, Kato M, Nakamura K, de Man SA, Cope H, Shashi V; Undiagnosed Diseases Network, Friedman J, Joset P, Steindl K, Rauch A, Muffels I, van Hasselt PM, Petit F, Smol T, Le Guyader G, Bilan F, Sorlin A, Vitobello A, Philippe C, van de Laar IMBH, van Slegtenhorst MA, Campeau PM, Au PYB, Nakashima M, Saitsu H, Yamamoto T, Nomura Y, Louie RJ, Lyons MJ, Dobson A, Plomp AS, Motazacker MM, Kaiser FJ, Timberlake AT, Fuchs SA, Depienne C*, Mignot C*. Missense and truncating variants in CHD5 in a dominant neurodevelopmental disorder with intellectual disability, behavioral disturbances, and epilepsy. Hum Genet. 2021 Jul;140(7):1109-1120. PMID: 33944996
Depienne C, Mandel JL. 30 years of repeat expansion disorders: What have we learned and what are the remaining challenges? Am J Hum Genet. 2021 May 6;108(5):764-785. PMID: 33811808
Mochel F, Rastetter A, Ceulemans B, Platzer K, Yang S, Shinde DN, Helbig KL, Lopergolo D, Mari F, Renieri A, Benetti E, Canitano R, Waisfisz Q, Plomp AS, Huisman SA, Wilson GN, Cathey SS, Louie RJ, Del Gaudio D, Waggoner D, Kacker S, Nugent KM, Roeder ER, Bruel AL, Thevenon J, Ehmke N, Horn D, Holtgrewe M, Kaiser FJ, Kamphausen SB, Abou Jamra R, Weckhuysen S, Dalle C, Depienne C. Variants in the SK2 channel gene (KCNN2) lead to dominant neurodevelopmental movement disorders. Brain. 2020 Dec 1;143(12):3564-3573. PMID: 33242881
Florian RT, Kraft F, Leitão E, Kaya S, Klebe S, Magnin E, van Rootselaar AF, Buratti J, Kühnel T, Schröder C, Giesselmann S, Tschernoster N, Altmueller J, Lamiral A, Keren B, Nava C, Bouteiller D, Forlani S, Jornea L, Kubica R, Ye T, Plassard D, Jost B, Meyer V, Deleuze JF, Delpu Y, Avarello MDM, Vijfhuizen LS, Rudolf G, Hirsch E, Kroes T, Reif PS, Rosenow F, Ganos C, Vidailhet M, Thivard L, Mathieu A, Bourgeron T, Kurth I, Rafehi H, Steenpass L, Horsthemke B; FAME consortium, LeGuern E, Klein KM, Labauge P, Bennett MF, Bahlo M, Gecz J, Corbett MA, Tijssen MAJ, van den Maagdenberg AMJM, Depienne C. Unstable TTTTA/TTTCA expansions in MARCH6 are associated with Familial Adult Myoclonic Epilepsy type 3. Nat Commun. 2019 Oct 29;10(1):4919. PMID: 3166403
Corbett MA, Kroes T, Veneziano L, Bennett MF, Florian R, Schneider AL, Coppola A, Licchetta L, Franceschetti S, Suppa A, Wenger A, Mei D, Pendziwiat M, Kaya S, Delledonne M, Straussberg R, Xumerle L, Regan B, Crompton D, van Rootselaar AF, Correll A, Catford R, Bisulli F, Chakraborty S, Baldassari S, Tinuper P, Barton K, Carswell S, Smith M, Berardelli A, Carroll R, Gardner A, Friend KL, Blatt I, Iacomino M, Di Bonaventura C, Striano S, Buratti J, Keren B, Nava C, Forlani S, Rudolf G, Hirsch E, Leguern E, Labauge P, Balestrini S, Sander JW, Afawi Z, Helbig I, Ishiura H, Tsuji S, Sisodiya SM, Casari G, Sadleir LG, van Coller R, Tijssen MAJ, Klein KM, van den Maagdenberg AMJM, Zara F, Guerrini R, Berkovic SF, Pippucci T, Canafoglia L, Bahlo M, Striano P, Scheffer IE, Brancati F, Depienne C, Gecz J. Intronic ATTTC repeat expansions in STARD7 in familial adult myoclonic epilepsy linked to chromosome 2. Nat Commun. 2019 Oct 29;10(1):4920. PMID: 31664034
Former group members
- Dr. rer. nat. Theresa Kühnel
- Lars Mohren
- Dr. med. Rahel Bettges, née Florian