
Clinical and molecular characterization of chromatinopathies
Description of the research group
Chromatinopathies represent a class of neurodevelopmental disorders caused by mutations in proteins responsible for chromatin remodeling and transcriptional regulation. The resulting dysregulation of gene expression favors the onset of a series of clinical features such as developmental delay, intellectual disability, facial dysmorphism, and behavioral disturbances.
Cornelia de Lange syndrome (CdLS) is a key example of a chromatinopathy. It is primarily caused by mutations affecting subunits or regulators of the cohesin complex. This complex is crucial for various molecular mechanisms, including sister chromatid cohesion, transcriptional regulation, and the formation of topologically associated domains. Interestingly, disease-causing variants in non-cohesin genes with overlapping functions have also been linked to CdLS. Many of these genes were previously associated with other distinct neurodevelopmental disorders that also fall under the spectrum of chromatinopathies, often considered as differential diagnoses for CdLS (Figure 1).
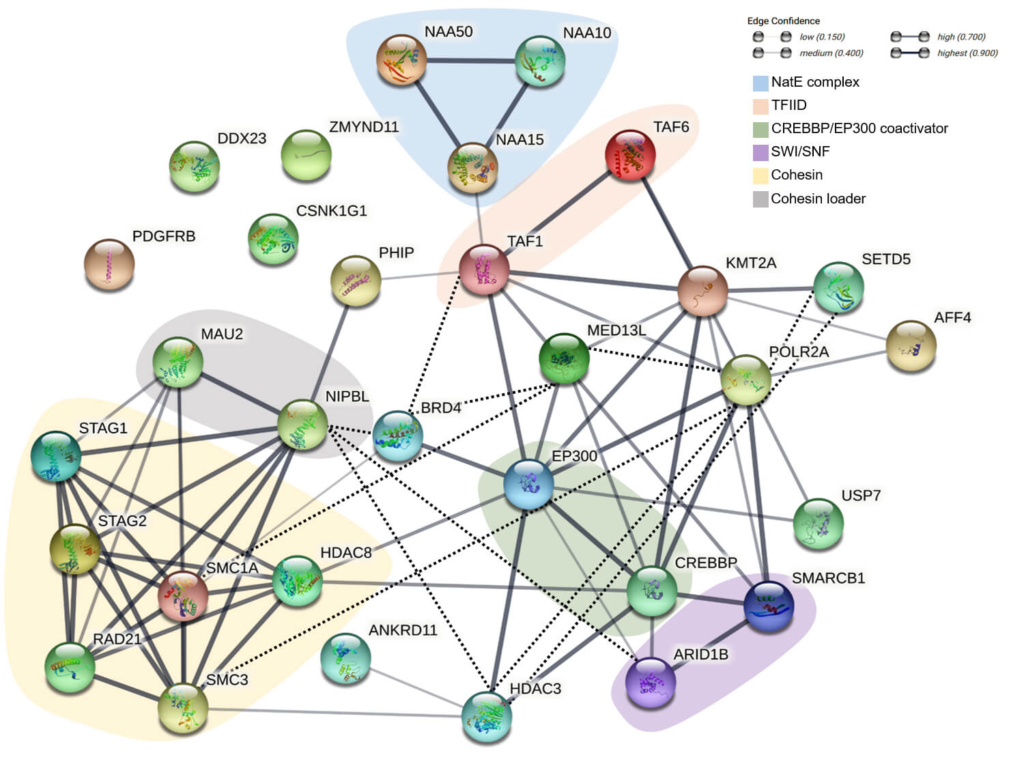
Figure 1. Diagram illustrating the functional and physical interactions within the chromatinopathies protein network. Source: Parenti and Kaiser, 2021. Front Neurosci, PMID: 34803598
In light of this, our research focus has expanded beyond CdLS to encompass other chromatin dysregulation disorders. We are dedicated to the clinical and molecular characterization of these conditions. Our goals include:
- Refining Genotype-Phenotype Correlations: We aim to deepen our understanding of the relationship between specific genetic mutations and their clinical manifestations, which is crucial for optimizing patient management strategies.
- Identifying Novel Disease Genes: We are dedicated to discovering new disease genes and describing the clinical features associated with their mutations, thereby enhancing the characterization of chromatinopathies.
- Delving into Molecular Mechanisms: Our research not only focuses on the genetic etiology and clinical aspects of chromatinopathies but also delves into the molecular mechanisms underlying these complex conditions. By characterizing these mechanisms, we hope to uncover new potential therapeutic targets.
Our ongoing projects include the clinical and molecular characterization of mutations in key genes such as MAU2, CDK16, AFF4, and CHD5. Through these initiatives, we hope to contribute to the broader understanding of chromatinopathies, ultimately improving outcomes for affected individuals.
Selected publications from the last 5 years
Parenti, I; Hesters, A; Gil-Salvador, M; Duffy, L; Kanber, D; Beygo, J; Kerkhof, J; Steenpaß, L; Leitão, E; Woestefeld, J; Boone, P M.; Kao, E M.; Alabdi, L; Aldhalaan, H M.; Alkuraya, F S.; Alshammari, M J.; Antonarakis, S E.; Basel, D; Cassinari, K; Polli Cellin, L de; Clause, A R.; Lima J, Alexander A de; Castro Leal, A de; Collins, S C.; Durand, B; Eckhold, J; Hashem, M O.; Jayakar, P; Khan, A O.; Kato, K; Kubica, R; Lyon, G J.; Marchi, E; McCarrier, J; Kimmig, L K.; Mizuno, S; Nicolas, G; Nishio, Y; Ogi, T; Pié, J; Prell, J; Puisac, B; Ramos, F J.; Ranza, E; Redin, C; Rush, E; Saitoh, S; Shamseldin, H E.; Starling, S; Astiazaran-Symonds, E; Taher, S; Kuechler, A; Sadikovic, B; Yalcin, B; Wendt, K S.; Kaiser, F J. (2025)
Pathogenic variants in the cohesin loader subunit MAU2 lead to a new Cornelia de Lange Syndrome subtype
Nat Commun. 2026 Mar 30;17(1):3036. doi: 10.1038/s41467-026-71177-6.
Moronta Gines, M; Wessels, M W.; Casa, V; van Staveren, T; Hof, A; Chung, W K.; Willems, M; Sandestig, A; Huening, I; Turnpenny, P; Lefebvre, M; Parenti, I; Kaiser, F J.; Demmers, J; van IJcken, W F. J.; Wendt, K S. (2025)
STAG2-truncating variants reveal a mosaic STAG2 inactivation pattern and compensatory mechanisms involving cohesin complex remodeling
iScience 28 (12), S. 114195. https://doi.org/10.1016/j.isci.2025.114195.
Sheppard SE, Bryant L, Wickramasekara RN, […], Parenti I, Kaiser FJ, Kuechler A, Busk ØL, Islam L, Siedlik JA, Henderson LB, Juusola J, Person R, Schnur RE, Vitobello A, Banka S, Bhoj EJ, Stessman HAF (2023)
Mechanism of KMT5B haploinsufficiency in neurodevelopment in humans and mice
Science advances, 9(10), eade1463. https://doi.org/10.1126/sciadv.ade1463
Kampmeier A, Leitão E, Parenti I, […], Kaiser FJ, Kuechler A. (2023)
PHIP-associated Chung-Jansen syndrome: Report of 23 new individuals
Frontiers in cell and developmental biology, 10, 1020609. https://doi.org/10.3389/fcell.2022.1020609
Parenti I, Leitão E, Kuechler A, […], Lesca G, Depienne C (2022)
The different clinical facets of SYN1-related neurodevelopmental disorders. Frontiers in cell and developmental biology, 10, 1019715. https://doi.org/10.3389/fcell.2022.1019715
Parenti I, Kaiser FJ (2021)
Cornelia de Lange Syndrome as Paradigm of Chromatinopathies
Frontiers in neuroscience, 15, 774950. https://doi.org/10.3389/fnins.2021.774950
Parenti I, Lehalle D, Nava C, […], Depienne C, Mignot C (2021)
Missense and truncating variants in CHD5 in a dominant neurodevelopmental disorder with intellectual disability, behavioral disturbances, and epilepsy
Human genetics, 140(7), 1109–1120. https://doi.org/10.1007/s00439-021-02283-2
Parenti I, Mallozzi MB, Hüning I, […], Deardorff MA, Gillessen-Kaesbach G, Kaiser FJ (2021)
ANKRD11 variants: KBG syndrome and beyond
Clinical genetics, 100(2), 187–200. https://doi.org/10.1111/cge.13977
Parenti I*, Rabaneda LG*, Schoen H, Novarino G (2020)
Neurodevelopmental Disorders: From Genetics to Functional Pathways
Trends in neurosciences, 43(8), 608–621. https://doi.org/10.1016/j.tins.2020.05.004
Parenti I, Diab F, Ruiz Gil S, Mulugeta E, Casa V, Berutti R, Brouwer RWW, Dupé V, Eckhold J, Graf E, Puisac B, Ramos F, Schwarzmayr T, van Staveren T, van IJcken WFJ, Strom TM, Pié J, Watrin E, Kaiser FJ, Wendt KS (2020)
MAU2 and NIPBL Variants Impair the Heterodimerization of the Cohesin Loader Subunits and Cause Cornelia de Lange Syndrome
Cell reports, 31(7), 107647. https://doi.org/10.1016/j.celrep.2020.107647
Avagliano L, Parenti I, Grazioli P, Di Fede E, Parodi C, Mariani M, Kaiser FJ, Selicorni A, Gervasini C, Massa V (2020)
Chromatinopathies: A focus on Cornelia de Lange syndrome
Clinical genetics, 97(1), 3–11. https://doi.org/10.1111/cge.13674
Fountain MD, Oleson D, Rech ME, […],, Parenti I, Kaiser FJ, Ehmke N, Schaaf CP (2019)
Pathogenic variants in USP7 cause a neurodevelopmental disorder with speech delays, altered behavior, and neurologic anomalies
Genetics in medicine : official journal of the American College of Medical Genetics, 21(8), 1797–1807. https://doi.org/10.1038/s41436-019-0433-1
Group Members
Dr. rer. nat.
Ilaria Parenti
Juliane Eckhold
M. Sc.
Alina Hesters
Prof. Dr. rer. nat.
Frank Kaiser
Contact person/contact details
Dr. rer. nat.
Ilaria Parenti
Molekulargenetik
Former group members
- Judith Eicker, née Schaffrath
- Nikita Schön